
Key Terms
FDA (Food and Drug Administration) is the United States agency responsible for protecting the public health by assuring the safety, efficacy, and security of drugs, biological products, and medical devices. Notably, they are not concerned with IP protection.
SAMD (Software as a Medical Device) is software that performs one or more medical functions; while the function may be embedded in a piece of hardware, the software itself performs the medical function.
AI (Artificial Intelligence) refers to data processes and algorithms that mimic human cognitive functions.
ML (Machine Learning) is a subset of artificial intelligence where the AI is able to learn from data and past experiences, using algorithms to analyze and interpret patterns in the data.
Timeline of FDA Considerations for AI/ML
April 2019: The FDA published an initial discussion paper regarding AI/ML-enabled SAMD.
January 2021: The FDA issued an action plan describing the strategy for AI/ML-enabled SAMD.
October 2022: The White House released a Blueprint for an AI Bill of Rights to discuss the design, use, and deployment of automated systems
September 2022: FDA releases report detailing the key findings from a Software Precertification Pilot Program, which included various shortcomings and informed changes needed to the initial proposed workflow.
December 2022: The Food and Drug Omnibus Reform Act of 2022, “FDORA,” added section 515C “Predetermined Change Control Plans [PCCP] for Devices” to the FD&C Act.
- Provides FDA with authority to approve or clear PCCPs for devices requiring premarket approval or premarket notification.
- i.e., changes consistent with an authorized PCCP will not require supplemental applications or new premarket notifications.
- FDA may require a PCCP to include:
- Labeling for safe and effective usage as the device changes pursuant to the plan.
- Notification requirements if the device does not function as intended.
- Performance requirements for changes made under the plan.
April 2023: FDA announces first AI/ML SAMD Draft Guidance (detailed here) with specific recommendations on PCCP content for submissions to the FDA.
October 2023: Executive Order is released on Safe, Secure, and Trustworthy Artificial Intelligence (AI)
Overview of Draft Guidance
Summary
The traditional regulatory framework for medical devices requires submissions to the FDA for review and approval before devices can enter the market. However, some machine-learning-based software uses data for continuous improvement, involving frequent updates that traditionally require multiple new marketing submissions.
In the April 2023 Draft Guidance, the FDA aims to address this iterative nature of AI/ML software development by proposing that manufacturers submit a Predetermined Change Control Plan to proactively seek premarket authorization for intended modifications that would otherwise require a new submission (see supplemental information).
This guidance applies to machine learning-enabled device software functions (ML-DSF) implemented automatically or manually (i.e., with human input).
Predetermined Change Control Plan (PCCP)
- Definition: the portion of a device’s marketing submission (510k, de novo, PMA) that pre-specifies intended modifications and describes their implementation protocol.
- Modifications included in a PCCP must maintain the device within intended use and the device’s indications for use (distinction in supplemental information).
- Once authorized, a PCCP is considered a technological characteristic of the approved device.
- Subsequent changes to an authorized PCCP will require a new marketing submission before implementation.
3 Required Components of a PCCP
- Description of Modifications: This section should enumerate specific planned modifications that can be verified and validated.
- Modification Protocol: This section details the implementation of modifications, including the verification and validation methods.
- Impact Assessment: This section (which can be standalone or integrated within the Modification Protocol) details the benefits and risks associated with implementing a PCCP, including a plan for risk mitigation.
510k Device Approvals if Predicate Device has PCCP
510k approval pathway is used when a new device has technological characteristics substantially equivalent to a predicate device.
When the predicate device includes an authorized PCCP, substantial equivalence determination must compare the new device to the predicate device’s version before changes are made under the PCCP
PCCP Required Components
A PCCP is required to have three components: Description of Modifications, Modification Protocol, and Impact Assessment. Below are high-level summaries of what content the FDA advises to include in each.
To assist with strategizing a PCCP, Appendix B of the Draft Guidance includes example scenarios of how a PCCP can be utilized.
1. Description of Modifications: Identify the specific, planned modifications to be implemented.
- Enumerated list of intended modifications with the rationale for each change
- Modifications must be within the device’s intended use and indications for use (distinction in supplemental information). Acceptable types of modifications include:
- Quantitative measures of ML-DSF performance specifications
- For instance, improvements to analytical performance by retraining the model with new data from the intended use population that falls within the same type and range of input signal.
- Device inputs to the ML-DSF
- For instance, expanding the algorithm to include new sources of the same signal type or limited modifications to accommodate new types of inputs.
- Device use and performance (albeit limited)
- For instance, authorizing a device for a specific subpopulation within the initially indicated population based on re-training the model on a more extensive data set specific to that subpopulation.
- Quantitative measures of ML-DSF performance specifications
- Each modification must be directly linked to a performance evaluation within the modification protocol to ensure that intended improvements are realized and validated.
2. Modification Protocol: Describe the methods that will be followed to implement each modification detailed in the Description of Modifications.
- Verification and Validation Methods: comprehensive explanation of the methods used to verify and validate the modifications. E.g., software testing, simulation, and field testing.
- Performance Evaluation Metrics: benchmarks of assessing performance post-modification.
- Implementation Steps: step-by-step procedure outlining the process from modification development to deployment, including code updates, integration testing, and transparent communication to users.
- Risk Management and Mitigation: identification of potential risks with modifications and strategies to mitigate them, including fallback procedures, monitoring systems, and contingency plans.
- Documentation: maintain comprehensive records of all processes to ensure traceability and accountability of each modification.
- Post-Implementation Review: a plan for reviewing the efficacy of modifications after implementation could include user feedback, analysis of performance data, and so on.
Appendix A of the Draft Guidance includes 70+ questions to assist with drafting a Modification Protocol.
3. Impact Assessment: Plan to assess and mitigate the benefits and risks of implementing a PCCP.
- Compare each modification implemented to the version without any modifications.
- Identify each modification’s benefits and risks, including risks of social harm.
- Plan to ensure the safety and effectiveness of the device.
- Describe how the implementation of a modification impacts the performance of another.
- Describe the collective impact of implementing all modifications.
- Example Elements:
- Safety Analysis
- Effectiveness Evaluation
- Usability Study
- Risk-Benefit Ratio
- Regulatory Impact
- Data Integrity and Privacy
- Post-Market Surveillance Plan
- Contingency Plan (e.g., rollback plans, user notifications, additional training, or support)
This section can stand alone or be incorporated into the Modification Protocol.
Supplemental Information
- Software Changes that Require Submission, source: Deciding When to Submit a 510(k) for a Software Change to an Existing Device
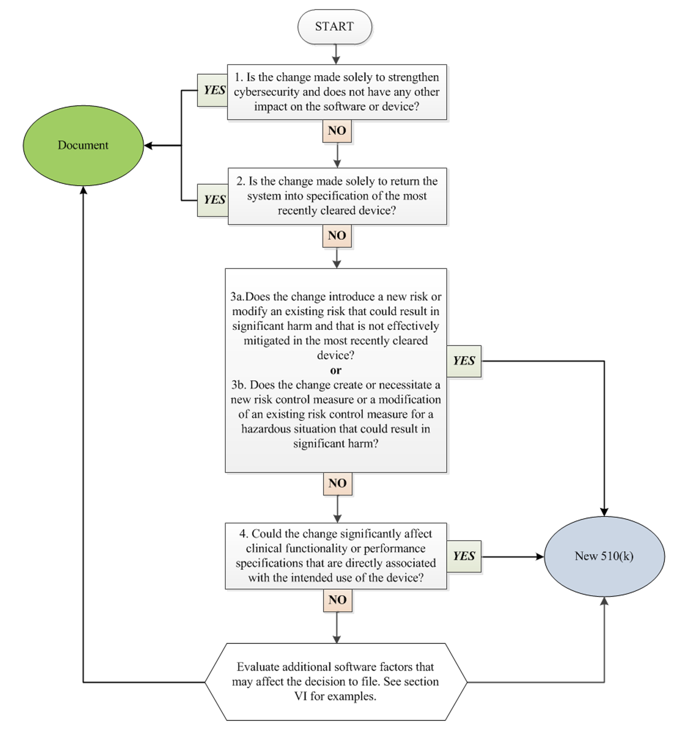
- Q-Submission process for obtaining FDA feedback on a proposed PCCP before submitting a marketing submission. For more information on the Q-Submission process, refer to the Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program guidance.
- Intended Use vs Indications for Use: Intended use is what you claim on your label that the device does. It’s the purpose of your device. Indications for use are the circumstances or conditions under which the device will be used, Source: Greenlight Guru Blog.
